Beta thalassemia is a common inherited blood disorder caused by mutations in the beta-globin gene, leading to reduced hemoglobin production and anemia1. The severity of the disease varies widely, ranging from mild symptoms in carriers to severe, life-threatening anemia requiring lifelong treatment2. Advances in diagnosis and management have improved outcomes, especially for patients with severe forms who depend on regular transfusions and iron chelation23.
Types of Beta Thalassemia
Beta thalassemia syndromes result from mutations in the HBB gene that impair beta-globin chain production, a key component of adult hemoglobin24. The clinical severity depends on the number and type of mutations inherited, leading to three main forms: minor, intermedia, and major2.
Beta Thalassemia Minor (Trait)
Beta thalassemia minor occurs when a person inherits one mutated beta-globin gene, typically resulting in a carrier state with mild or no symptoms2. Carriers usually have mild anemia due to ineffective red blood cell production but do not require treatment2. This form is often diagnosed incidentally during routine mcv blood test results meaning and normal range tests and does not affect life expectancy23.
Beta Thalassemia Intermedia
Beta thalassemia intermedia arises from inheriting two mutated beta-globin genes that cause a moderate reduction in beta-globin synthesis2. Patients experience variable anemia severity, often presenting in childhood or adolescence23. They may require intermittent blood transfusions depending on clinical status2. Complications such as bone deformities and enlarged spleen are common due to chronic anemia and marrow expansion2.
Beta Thalassemia Major (Cooley's Anemia)
Beta thalassemia major, also known as Cooley's anemia, results from homozygous or compound heterozygous mutations causing absent or severely reduced beta-globin production23. Symptoms typically appear within the first two years of life and include severe anemia, failure to thrive, and jaundice25. Patients require lifelong regular blood transfusions and iron chelation therapy to survive23. Without treatment, complications such as organ enlargement and bone deformities develop6.
Beta Thalassemia Symptoms
The symptoms of beta thalassemia vary according to the disease type and genetic severity, ranging from none to severe anemia and organ complications24.
Beta Thalassemia Minor Symptoms
Individuals with beta thalassemia minor are often asymptomatic and may only have mild anemia2. Common mild symptoms include fatigue and weakness due to reduced red blood cell production and hemolysis26. Most carriers lead normal lives without requiring treatment2.
Beta Thalassemia Intermedia Symptoms
Symptoms usually appear in childhood or adolescence with moderate anemia2. Fatigue is common, along with jaundice caused by increased red blood cell breakdown23. Patients often develop hepatosplenomegaly (enlarged liver and spleen) and bone changes due to marrow expansion27. Other complications include slow growth and leg ulcers28.
Beta Thalassemia Major Symptoms
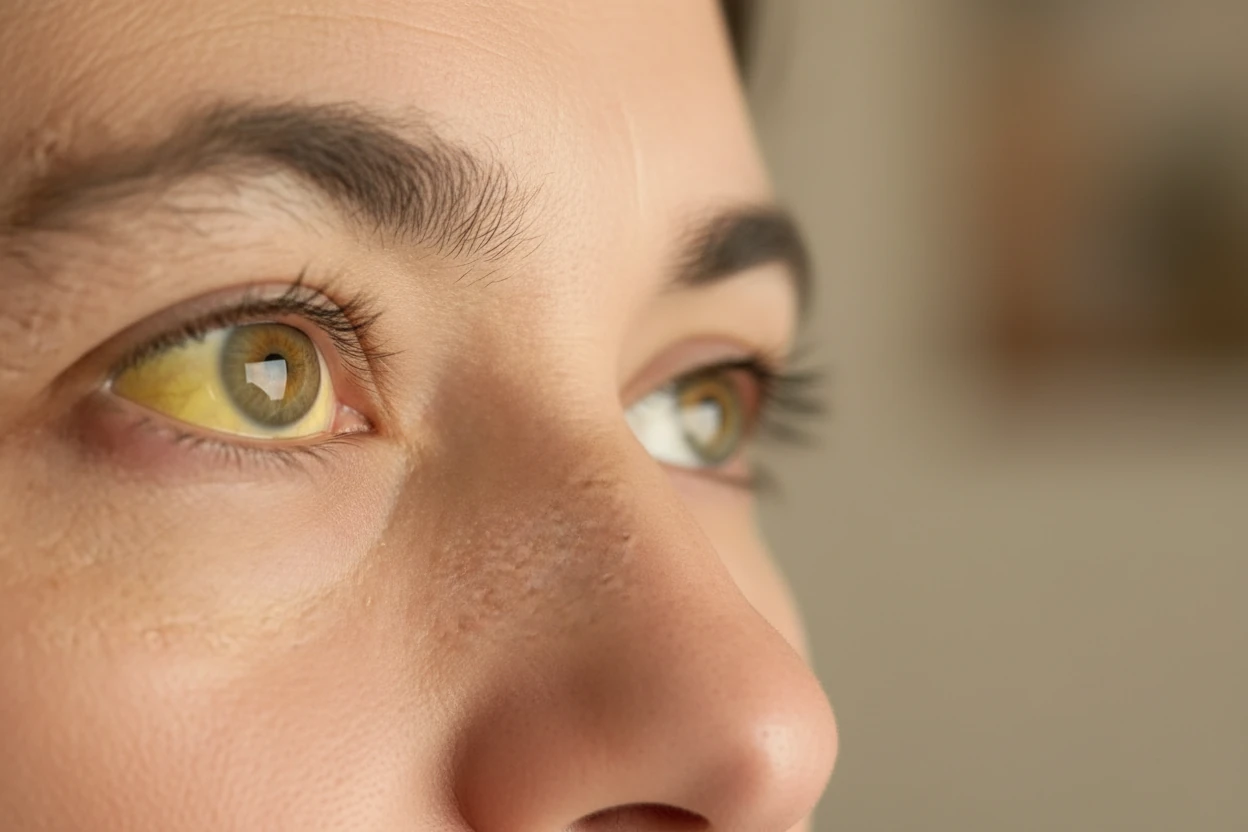
Severe anemia manifests early in infancy with symptoms such as pale skin, poor appetite, irritability, and frequent infections265. Failure to thrive and abdominal swelling from enlarged organs are common2. Bone deformities develop due to marrow overactivity, and untreated patients face life-threatening complications23. Iron overload from transfusions can cause heart failure and endocrine problems25.
Causes and Risk Factors
Beta thalassemia is a hereditary genetic disorder inherited in an autosomal recessive pattern24. The HBB gene on chromosome 11 encodes the beta-globin chain of hemoglobin, essential for oxygen transport in red blood cells21. Mutations reduce or abolish beta-globin synthesis, leading to ineffective erythropoiesis (red blood cell production) and anemia29.
Both parents must be carriers of beta thalassemia mutations for a child to inherit the disease2. The inheritance risks are:
- 25% chance of inheriting two mutated genes and developing beta thalassemia disease
- 50% chance of inheriting one mutated gene and becoming a carrier
- 25% chance of inheriting two normal genes and being unaffected21
Risk Factors
- Family history of beta thalassemia or carrier status2
- Ethnic background, especially Mediterranean, Middle Eastern, Central and Southeast Asian, and Indian subcontinent populations25
- Consanguineous marriages increasing the likelihood of inheriting mutations2
- Lack of genetic counseling or screening in at-risk populations2
Diagnosis and Testing
Beta thalassemia diagnosis involves clinical evaluation and laboratory testing, including blood counts and genetic analysis23. Diagnosis can occur at any age depending on symptom presentation2.
Key diagnostic methods include:
- Complete blood count (CBC) showing anemia and microcytosis (small red blood cells)210
- Hemoglobin electrophoresis to detect abnormal hemoglobin patterns such as elevated HbA2 and HbF27
- Genetic testing to identify mutations in the HBB gene and assess disease severity21
- Prenatal diagnosis through chorionic villus sampling (CVS) or amniocentesis if both parents are carriers21
- Non-invasive prenatal testing (NIPT) can screen early in pregnancy but requires confirmation by invasive testing111
Risk assessment includes evaluating ethnicity, family history, and clinical signs to guide testing2.
Beta Thalassemia Treatment Options
Treatment aims to correct anemia, prevent complications, and improve quality of life23. The approach depends on disease severity.
Blood Transfusions
Regular blood transfusions are the mainstay for beta thalassemia major and some intermedia cases23. Transfusions increase hemoglobin levels and oxygen delivery by supplementing red blood cells2. Frequency varies:
- Beta thalassemia major patients typically receive transfusions every 3 to 4 weeks2
- Beta thalassemia intermedia patients may need transfusions intermittently during illness or surgery2
Transfusions improve growth, development, and survival but carry risks of iron overload2.
Iron Chelation Therapy
Iron overload from chronic transfusions can damage organs like the liver, heart, and endocrine glands212. Chelation therapy removes excess iron to prevent toxicity2. Common iron chelators include:
- Deferoxamine (injected)2
- Deferiprone (oral)2
- Deferasirox (oral)2
Chelation is essential for transfusion-dependent patients to reduce morbidity and mortality23.
Blood and Bone Marrow Transplant
Hematopoietic stem cell transplant (bone marrow transplant) is the only known cure for severe beta thalassemia35. It replaces defective blood-forming cells with healthy donor cells, potentially eliminating the need for transfusions and chelation5. Transplant success depends on donor availability and patient condition2.
Other Treatments
- Folic acid supplements support red blood cell production3
- Splenectomy (surgical removal of the spleen) may be considered in some cases to reduce anemia and transfusion needs26
- Emerging therapies like luspatercept aim to reduce transfusion dependence by improving red blood cell production3
- Regular monitoring of heart and liver function is critical to manage complications213
Prevention and Genetic Counseling
Beta thalassemia cannot be prevented, but genetic counseling can reduce disease incidence by informing at-risk families214. Carrier screening is recommended for individuals with family history or from high-prevalence ethnic groups2. Genetic counseling helps families understand inheritance patterns and reproductive options2.
Preimplantation genetic diagnosis (PGD) during in vitro fertilization (IVF) can identify embryos without beta thalassemia mutations before implantation, preventing disease transmission2.
Related Blood Disorders
Beta thalassemia is associated with several complications affecting multiple organs due to chronic anemia and iron overload212.
- Iron overload causes liver and heart toxicity, leading to organ failure215
- Endocrine dysfunctions such as diabetes, hypothyroidism, and hypogonadism are common212
- Bone disease results from marrow expansion and mineral loss, increasing fracture risk216
- Cardiac complications from iron overload are a leading cause of death in beta thalassemia217
- Chronic anemia and poor circulation can cause leg ulcers and delayed growth28
Living With Beta Thalassemia
Living with beta thalassemia requires ongoing medical care and lifestyle adjustments to manage symptoms and prevent complications213. Regular transfusions and chelation therapy impact daily routines but improve quality of life and survival23. Support systems, including healthcare teams and patient communities, are vital for emotional and physical well-being13.
With proper treatment, many individuals with beta thalassemia can lead fulfilling lives and reach middle age or beyond2. Adherence to therapy and monitoring are essential to reduce complications and improve outcomes2.
“It gives you so much more freedom when you’re being honest and not trying to hide the truth of your experience and who you are.”
— Josephine, bluebird bio patient community13
Frequently Asked Questions
What causes beta thalassemia?
Beta thalassemia is caused by inherited mutations in the HBB gene that reduce or eliminate beta-globin production, leading to anemia24.
Can beta thalassemia be cured?
The only known cure is a bone marrow or stem cell transplant, which replaces defective blood cells with healthy ones35.
Do all patients need blood transfusions?
Not all. Beta thalassemia minor usually does not require treatment, while intermedia patients may need transfusions occasionally. Major patients require regular transfusions23.
Why is iron chelation therapy important?
Chronic transfusions cause iron buildup that damages organs. Chelation removes excess iron to prevent complications212.
Can beta thalassemia be prevented?
While the disease cannot be prevented, genetic counseling and carrier screening can reduce the risk of affected children2.