Thalassemia is an inherited blood disorder that affects the body’s ability to produce normal hemoglobin, the protein in red blood cells responsible for carrying oxygen throughout the body1. This condition leads to fewer healthy red blood cells and varying degrees of anemia, ranging from mild to severe1. Thalassemia is more common in people with ancestry from regions where malaria is or was prevalent, such as parts of Asia, Africa, and the Mediterranean2. Understanding the types, symptoms, genetic causes, diagnosis, and treatment options is essential for managing this lifelong condition3.
Types of Thalassemia
Thalassemia is classified based on which globin chain of hemoglobin is affected: alpha or beta. Hemoglobin consists of two alpha and two beta globin chains, and mutations in the genes encoding these chains cause the disorder3.
Alpha Thalassemia
Alpha thalassemia results from deletions or mutations in the HBA1 and HBA2 genes located on chromosome 16, which encode the alpha globin chains of hemoglobin34. Each person inherits four alpha globin genes, two from each parent4. The clinical severity depends on how many of these genes are deleted or mutated:
- Silent Carrier: One alpha globin gene deletion, usually no symptoms4.
- Alpha Thalassemia Trait: Two gene deletions, causing mild or no symptoms4.
- Hemoglobin H Disease: Three gene deletions, leading to moderate to severe anemia45.
- Alpha Thalassemia Major (Hydrops Fetalis): All four alpha genes deleted, usually fatal before or shortly after birth45.
The absence or dysfunction of alpha globin chains causes an excess of beta or gamma globin tetramers, which are nonfunctional and damage red blood cells5.
Beta Thalassemia
Beta thalassemia is caused by mutations in the HBB gene on chromosome 11, which encodes the beta globin chains of hemoglobin36. Unlike alpha thalassemia, beta thalassemia mutations are mostly point mutations leading to reduced or absent beta globin production6. The severity depends on whether one or both beta globin genes are affected:
- Beta Thalassemia Trait (Carrier): One mutated gene, usually mild or no symptoms2.
- Beta Thalassemia Intermedia: Variable severity, with moderate anemia6.
- Beta Thalassemia Major: Two mutated genes, causing severe anemia requiring lifelong transfusions67.
Beta thalassemia major patients often present with severe anemia, jaundice, growth retardation, and bone deformities early in life7.
Thalassemia Symptoms and Signs
Symptoms of thalassemia vary widely depending on the type and severity of the disorder. Mild forms may cause no symptoms, while severe forms can lead to significant health problems3.
Common symptoms include:
- Fatigue and weakness due to anemia18.
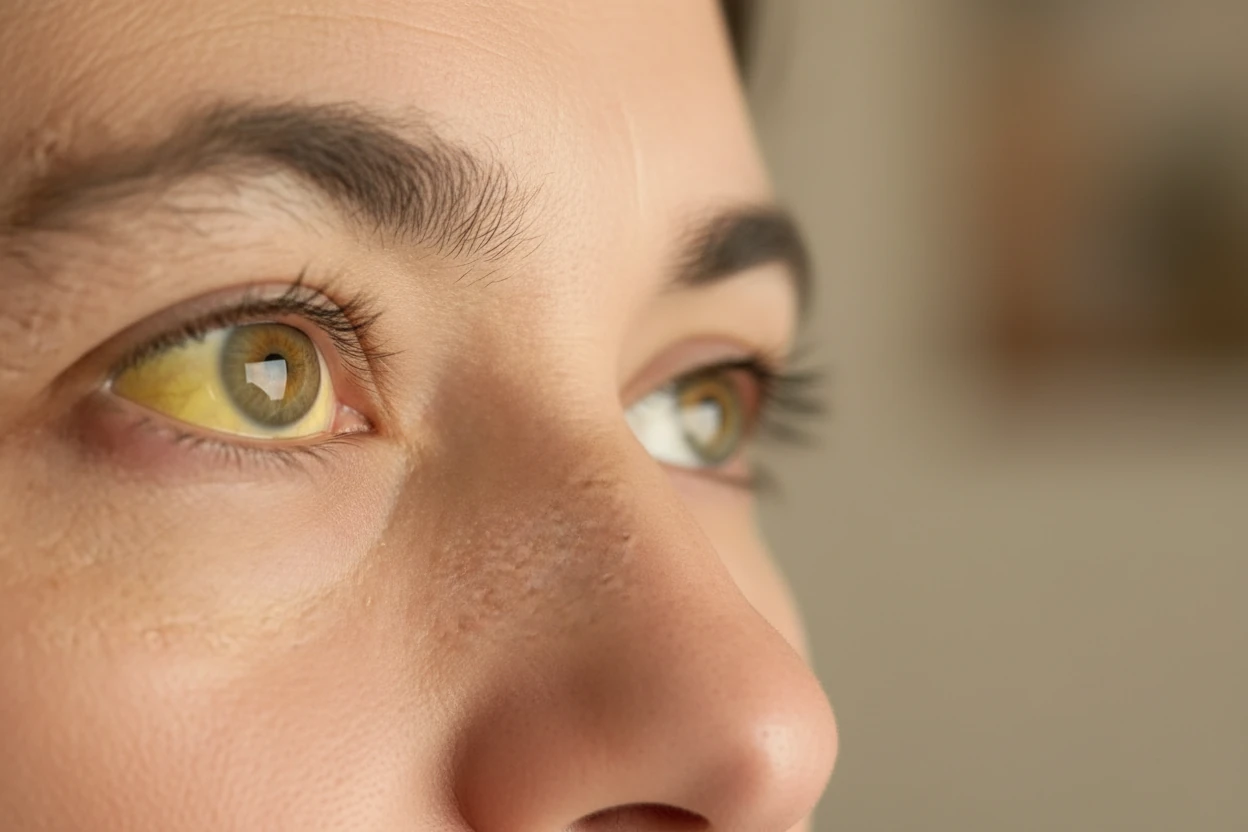
- Pale or yellowish skin (jaundice) caused by hemolysis (red blood cell breakdown)62.
- Shortness of breath and dizziness from low oxygen delivery1.
- Poor appetite and delayed growth in children92.
- Bone deformities, especially in the face and skull, due to bone marrow expansion101112.
- Enlarged spleen (splenomegaly) from increased red blood cell destruction6.
- Dark or tea-colored urine from hemolysis byproducts2.
Severe thalassemia types typically manifest symptoms within the first two years of life613. Nutritional deficiencies, including low levels of vitamins C, D, folate, zinc, and copper, are common and contribute to complications6.
Genetic Causes and Inheritance
Thalassemia is caused by inherited mutations in the globin genes that disrupt normal hemoglobin synthesis3. The disorder follows an autosomal recessive inheritance pattern, meaning a child must inherit defective genes from both parents to be affected4.
- Alpha thalassemia results from deletions or mutations in the HBA1 and HBA2 genes on chromosome 164.
- Beta thalassemia arises from mutations in the HBB gene on chromosome 116.
- Carriers inherit one mutated gene and usually do not show symptoms but can pass the gene to their children3.
- If both parents are carriers, each pregnancy has a 25% chance of producing a child with thalassemia4.
Risk Factors
- Family history of thalassemia or carrier status increases risk3.
- Ethnic background plays a role, with higher prevalence in Mediterranean, Middle Eastern, South Asian, African, and Southeast Asian populations214.
- Consanguineous marriages may increase the risk of inheriting thalassemia3.
Diagnosis and Testing Methods
Diagnosing thalassemia involves a combination of clinical evaluation, family history, and laboratory testing3.
- Complete Blood Count (CBC): Detects anemia and shows microcytic (small) and hypochromic (pale) red blood cells typical of thalassemia615.
- Peripheral Blood Smear: Reveals characteristic red blood cell abnormalities such as microcytosis and fragmentation15.
- Hemoglobin Electrophoresis: Differentiates thalassemia from other hemoglobin disorders by identifying abnormal hemoglobin variants6.
- Genetic Testing: Confirms specific gene mutations in HBA1, HBA2, or HBB genes4.
- Prenatal Diagnosis: Chorionic villus sampling (CVS) or amniocentesis can detect thalassemia mutations early in at-risk pregnancies413.
Early diagnosis allows for timely management and genetic counseling for families4.
Treatment Options and Management
Treatment depends on the type and severity of thalassemia, ranging from no treatment for mild cases to intensive therapies for severe forms3.
Blood Transfusions
Regular blood transfusions are the main treatment for moderate to severe thalassemia, especially beta thalassemia major616.
- Transfusions provide healthy red blood cells to alleviate anemia and improve oxygen delivery36.
- Frequency varies by severity; severe cases may require transfusions every 3–4 weeks6.
- Transfusions reduce symptoms such as fatigue and improve growth and development in children6.
Iron Chelation Therapy
Chronic transfusions cause iron overload, a major complication that can damage organs like the heart, liver, and endocrine glands616.
- Chelation therapy uses medications such as deferoxamine, deferiprone, or deferasirox to remove excess iron from the body6.
- Chelation is essential to prevent organ damage and improve long-term outcomes6.
- Monitoring iron levels regularly guides chelation treatment6.
Bone Marrow Transplant
Bone marrow or stem cell transplant is currently the only curative treatment for thalassemia716.
- Transplant involves replacing the patient’s defective bone marrow with healthy stem cells from a compatible donor, often a sibling7.
- It eliminates the need for lifelong transfusions and chelation7.
- Transplant success depends on donor compatibility and patient condition16.
Other Treatments
- Folic acid supplements support red blood cell production and are often prescribed alongside other treatments16.
- New therapies like luspatercept, an injection that stimulates red blood cell production, offer additional options for some patients13.
- Gene therapy is an emerging treatment involving modification of the patient’s own stem cells to correct the genetic defect1718.
- Splenectomy (removal of the spleen) may be considered if the spleen enlarges significantly and increases transfusion needs7.
“Thalassemia is an inherited blood disorder that affects the body's ability to produce normal hemoglobin.”
— Rabi Hanna, Cleveland Clinic1
Thalassemia Prevention Strategies
Thalassemia cannot be prevented because it is inherited genetically3. However, prevention focuses on:
- Carrier Screening: Identifying carriers, especially in high-risk populations or families with a history of thalassemia34.
- Genetic Counseling: Providing risk assessment and reproductive options for carrier couples to make informed decisions3.
- Prenatal Testing: Early detection of affected fetuses through CVS or amniocentesis allows families to prepare or consider options4.
- Advances in noninvasive prenatal testing and preimplantation genetic diagnosis offer future preventive possibilities419.
Related Blood Disorders
Thalassemia is part of a group of inherited hemoglobin disorders known as hemoglobinopathies, which also include sickle cell disease20. These disorders share features such as defective hemoglobin synthesis and anemia but differ in genetic causes and clinical manifestations20. Understanding related conditions helps guide diagnosis and treatment strategies3.
Living With Thalassemia
Living with thalassemia involves managing both the disease and its complications3.
- Iron Overload: Excess iron from transfusions accumulates in organs, causing heart disease, liver damage, and endocrine problems like diabetes and growth delays621.
- Bone Disease: Bone marrow expansion leads to skeletal deformities, osteoporosis, and increased fracture risk36.
- Splenomegaly: Enlarged spleen increases infection risk and may impair immune function36.
- Infections: Patients are immunocompromised and require vaccinations and infection prevention measures3.
- Psychosocial Support: Chronic treatment and frequent hospital visits impact quality of life, making support systems essential22.
Adherence to treatment and healthy lifestyle choices improve outcomes and life expectancy36.
💡 Did You Know?
Thalassemia is an inherited blood disorder that affects your body's ability to produce hemoglobin and healthy red blood cells. Types include alpha and beta thalassemia.1
Thalassemia FAQs
Q: Can thalassemia be cured?
A: Bone marrow or stem cell transplant is currently the only cure for thalassemia, but it requires a compatible donor and carries risks. Gene therapy is an emerging option718.
Q: How is thalassemia inherited?
A: Thalassemia follows an autosomal recessive pattern. A child must inherit mutated genes from both parents to have the disease. Carriers usually do not show symptoms but can pass the gene to their children43.
Q: What are the main treatments for severe thalassemia?
A: Regular blood transfusions and iron chelation therapy are the main treatments. New drugs and bone marrow transplant may also be options616.
Q: Can people with thalassemia live normal lives?
A: With proper treatment and management, many people with mild to moderate thalassemia live normal or near-normal lives. Severe cases require lifelong care63.